The paper explores the challenges of meta-analysis, from data collection and preprocessing to statistical analysis and result interpretation. The author emphasizes the necessity for continuous improvement in this field, highlighting its complex, multifaceted nature. This scientific article will be useful for researchers, educators, and students, as well as a wide audience interested in the current state of biomedical research in genetics.
Introduction
Meta-analysis of associations of single nucleotide polymorphisms (SNPs) in genes with various diseases is one of the key methods of genetic epidemiology. Meta-analysis is also an essential step in developing any drug targeting diseases caused by single nucleotide polymorphisms. However, conducting this analysis requires significant time and human resources for processing large datasets, integrating them from different sources, and performing statistical analysis. To address this problem and improve the efficiency of meta-analyses, a program was developed to automate key stages, accelerating the process and reducing the likelihood of errors.
The primary goal of this program is to automate the entire cycle of SNP-disease association meta-analysis, from data collection and filtering to final statistical processing and results visualization. Implementing this program significantly reduces research time, eases the workload on research teams, and minimizes human error—especially crucial when working with large datasets. By employing automated algorithms for routine tasks, researchers can focus on interpreting and applying results in clinical practice, opening new opportunities to understand the genetic basis of diseases.
Meta-Analysis and Terminology
Meta-Analysis
Meta-analysis refers to a scientific methodology that combines results from multiple studies using statistical methods to test one or more related scientific hypotheses. Simply put, meta-analysis identifies a “mean” value to trust from all results previously obtained by other researchers. In our case, this involves the association of an SNP in a gene with a disease. For example, we performed a meta-analysis to test the association of rs7903146 in the TCF7L2 gene with type 2 diabetes [1].
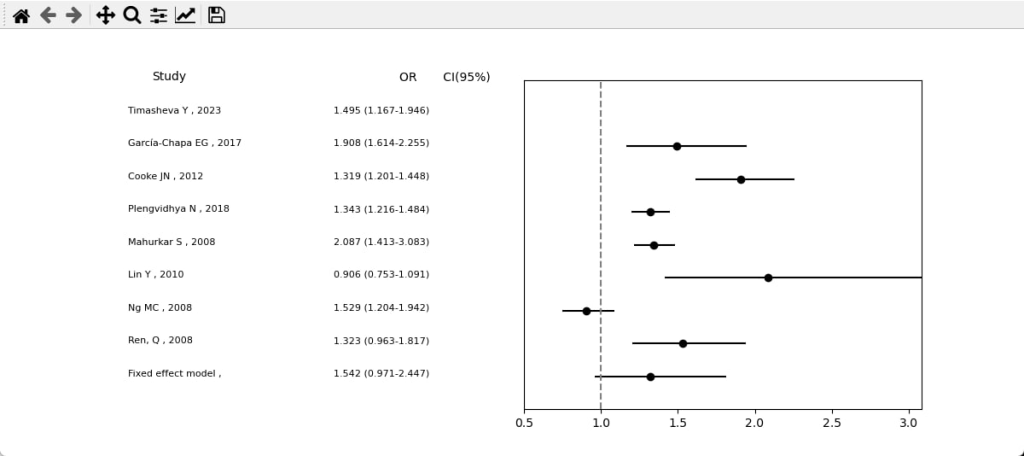
Example of Visualization Results from Genetic Meta-Analysis Fig. 1
Single Nucleotide Polymorphism (SNP)
SNPs are variations in DNA sequences involving a single nucleotide in the genome of individuals of the same species or between homologous regions of homologous chromosomes.

Example of Single Nucleotide Polymorphism Fig. 2
Minor Allele Frequency (MAF)
MAF is the frequency at which the second most common allele occurs in a population. These frequencies play a surprising role in heritability because MAF variants that occur only once, known as «singletons,» drive a large amount of selection.
Genome-Wide Association Studies (GWAS)
GWAS refers to a field of biological (often biomedical) research investigating associations between genomic variants and phenotypic traits. Often, GWAS focuses on identifying connections between SNPs and human diseases, though the term can also apply to other organisms.
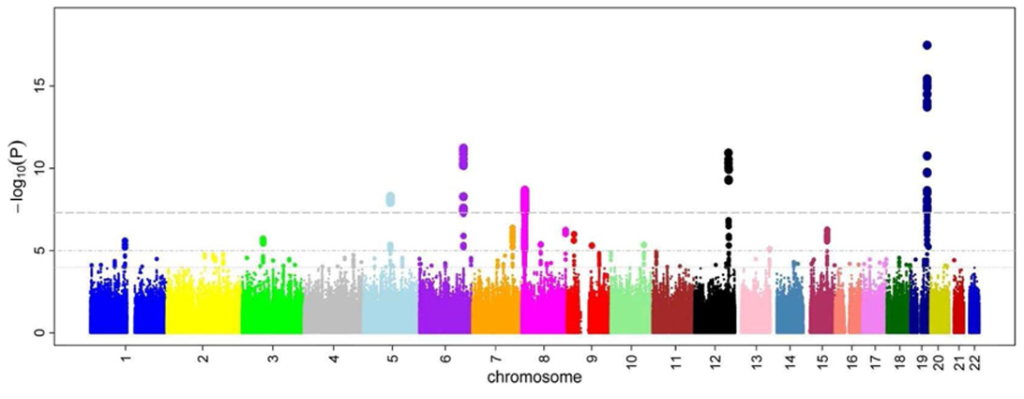
Manhattan plot depicting some closely related risk loci (GWAS method) Fig. 3
Statistics
The drug development process is a multi-stage and lengthy procedure, taking from 1 to 5 or even 10 years. These extended timelines pose a critical risk for patients with SNP-associated diseases, who may die before effective treatments are available. Conducting meta-analyses alone can take weeks or months [2]. It’s essential to note that many SNP-related diseases lack therapeutic drugs, and their number far exceeds diseases with available treatments.
Challenges
Traditional approaches to genetic meta-analysis for identifying SNP-disease associations have significant limitations. The process can take weeks or months, delaying results and negatively impacting clinical outcomes. These delays lower patients’ chances of receiving timely personalized therapy. Extended time frames also reduce the development of genetic-based treatments due to the substantial time required for creating and validating associations.
Additionally, manual analysis increases the risk of errors due to human factors. Researchers conducting meta-analyses must handle routine analytical tasks, contributing to professional burnout and decreased productivity. This high workload slows research processes and detracts from primary scientific activities, hindering the pace of developing new treatments.
Core Concept: Solving the Problem
The project involves developing a program for fully automating the process of genetic meta-analysis of SNP-disease associations. The program encompasses all research stages, from data collection and preprocessing to statistical analysis and result interpretation. The goal is to reduce analysis time, minimize human errors, and optimize research teams’ workflows. Implementing this program significantly accelerates the identification of genetic associations and simplifies drug development for SNP-related diseases.
Program Structure
The program includes six key stages. The process begins with the user entering specific keywords in the web interface and ends with obtaining results.
- User Input:
The researcher specifies:- SNP (e.g., rs7903146)
- Gene containing it (TCF7L2)
- Associated disease names (e.g., Type 2 Diabetes, T2D).
- Article Retrieval:
Based on these keywords, the program searches PubMed and retrieves relevant articles. Abstracts are scanned for gene, SNP, and disease mentions, and information is categorized. - Data Parsing:
Articles with full text are analyzed for terms like «MAF» and «GWAS» [3]. Data is sorted into categories for further meta-analysis. - Data Extraction:
Tables and accompanying text are processed, extracting parameters like cohort size, MAF, and GWAS statistics using GPT-4 [4]. - Meta-Analysis:
Extracted datasets are processed in Python to conduct two meta-analysis types, visualized via Forest plots, and calculate key metrics like p-value and Odds Ratio. - Result Presentation:
Results are returned to the user interface for easy interpretation and further use.
Conclusion
The program is currently functional but requires refinements, including enhanced article filtering, improved parsing, and interface updates. Upon completion, it will be ready for medical application, accelerating research and saving lives.
Statistical Impact
The program reduces meta-analysis time to ~5 minutes, making it approximately 45,500 times faster than manual methods. This time-saving increases patient survival rates by ~2.34%, equating to saving critical months in clinical practice.
References
1. Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, Nejjari C, Patsch W, Chikri M, Meyre D, Froguel P. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med (Berl). 2007 Jul;85(7):777-82. doi: 10.1007/s00109-007-0203-4. Epub 2007 May 3. PMID: 17476472.2. Zhang H, Deng L, Schiffman M, Qin J, Yu K (2020). "Generalized integration model for improved statistical inference by leveraging external summary data". Biometrika. 107 (3): 689–703
3. Giorgi, Federico M.; Ceraolo, Carmine; Mercatelli, Daniele (27 April 2022). "The R Language: An Engine for Bioinformatics and Data Science". Life. 12 (5): 648. Bibcode:2022Life...12..648G. doi:10.3390/life12050648. PMC 9148156. PMID 35629316
4. Bagde H, Dhopte A, Alam MK, Basri R. A systematic review and meta-analysis on ChatGPT and its utilization in medical and dental research. Heliyon. 2023 Nov 29;9(12):e23050. doi: 10.1016/j.heliyon.2023.e23050. PMID: 38144348; PMCID: PMC10746423.