Abstract: The article presents an analysis of current concepts on the molecular mechanisms of apoptosis – programmed cell death that is crucial for maintaining tissue homeostasis and the development of many diseases. The main pathways of apoptosis activation are considered: the extrinsic (receptor-mediated) and intrinsic (mitochondrial) pathways. The role of lipid mediators, primarily ceramide, in triggering proapoptotic signals through modulation of protein kinase activity and ion channels is shown. Special attention is paid to the caspase cascade as the central executive mechanism, as well as to the crosstalk between extrinsic and intrinsic pathways involving the Bid protein. The significance of ion channels, particularly potassium channels, in the regulation of membrane potential and their involvement in apoptotic signaling is highlighted. Based on the analysis, it is concluded that apoptosis is a complex integrated system combining biochemical and biophysical mechanisms. Understanding these processes opens prospects for the development of targeted therapeutic approaches in cancer, autoimmune and neurodegenerative diseases.
Keywords: apoptosis, caspases, ceramide, ion channels, mitochondrial pathway, death receptors, signal integration
Введение
Апоптоз представляет собой строго регулируемый процесс программируемой клеточной гибели, обеспечивающий баланс между пролиферацией и смертью клеток, необходимый для нормального функционирования тканей и предотвращения патологий. Регуляция апоптоза осуществляется на молекулярном уровне с участием сигнальных молекул, ферментов и ионных каналов.
Ключевая роль принадлежит липидным медиаторам, прежде всего церамиду — биологически активному сфинголипиду, образующемуся в ответ на стрессовые сигналы. Церамид активирует протеинкиназы (в том числе MAPK), что запускает внутриклеточные каскады, направленные на активацию генов, ответственных за апоптоз.
Ионные каналы, изменяя ионный баланс клетки, участвуют в регуляции апоптоза. Липидные медиаторы модулируют активность каналов, контролируя поток ионов (кальций, калий), что ведёт к изменению мембранного потенциала и запуску каспазного каскада. Повышение внутриклеточного кальция служит сигналом для активации протеаз, расщепляющих ключевые белки.
Центральным механизмом апоптоза является каспазный каскад — последовательная активация цистеиновых протеаз. Каспазы подразделяются на инициаторные (активируются первыми) и эффекторные (отвечают за деградацию структурных и регуляторных белков). Активация каспаз контролируется двумя основными путями: внешним и внутренним.
Внешний путь инициируется связыванием лигандов с рецепторами смерти на мембране клетки, что приводит к формированию сигнального комплекса и активации каспаз-8 или -10. Внутренний путь запускается внутриклеточными стрессорами (повреждение ДНК, нарушение митохондриальной мембраны), вызывая выход проапоптотических факторов, в том числе цитохрома c, из митохондрий. Цитохром c связывается с белком апап-1, активируя каспазу-9 и последующий каспазный каскад.
Данная работа посвящена изучению взаимодействия указанных молекулярных компонентов и их влияния на активацию апоптоза. Рассматриваются механизмы модуляции ионных каналов, обеспечивающие дополнительные уровни регуляции. Особое внимание уделяется комплексной интеграции сигналов внешнего и внутреннего путей, обеспечивающей точность и эффективность программы клеточной смерти.
Полученные результаты важны для углубления понимания молекулярных механизмов апоптоза и разработки перспективных терапевтических подходов к лечению заболеваний, связанных с нарушением клеточной гибели (рак, нейродегенеративные и аутоиммунные патологии).
- Перспективы исследования молекулярных механизмов апоптоза
Современные исследования молекулярных механизмов апоптоза направлены на выявление новых элементов регуляции, понимание междисциплинарных взаимодействий и разработку методов точного контроля этого процесса в живых организмах. Несмотря на прогресс в изучении классических путей (митохондриального и рецепторного), остаётся необходимость в более глубоком понимании динамики сигнальных сетей и тонких молекулярных взаимодействий, определяющих судьбу клетки.
Развиваются интегративные подходы, сочетающие молекулярную биологию с методами in vivo визуализации (ПЭТ, МРТ, флуоресцентный имиджинг), что позволяет отслеживать апоптоз в реальном времени в тканево-специфическом контексте. Активно создаются специализированные контрастные агенты для неинвазивного мониторинга клеточной гибели.
Важным направлением является изучение вариабельности апоптоза в различных клеточных типах, особенно в иммунных клетках и микроокружении опухолей. Изменения чувствительности лимфоцитов к проапоптотическим стимулам остаются в фокусе исследований.
С терапевтической точки зрения, модификация апоптоза становится приоритетом персонализированной медицины. Использование апоптоз-модулирующих агентов (мелатонин, BH3-миметики), а также воздействие на ионные каналы и липидные медиаторы позволяют тонко настраивать клеточный ответ. Перспективны комбинированные подходы, направленные на инициацию гибели патологических клеток и преодоление резистентности к традиционной терапии.
Апоптоз представляет собой результат взаимодействия сложной сети сигналов, включающей внешние рецепторы, митохондриальные сигналы, биоэлектрические изменения мембран и транскрипционные факторы. Такой комплексный характер требует системного подхода в исследованиях и клинической практике.
- Основы регуляции апоптоза на клеточном уровне
Апоптоз — генетически запрограммированный процесс клеточной гибели, при котором клетка распадается на апоптотические тельца, фагоцитируемые макрофагами без воспалительного ответа [4]. Этот механизм обеспечивает удаление ненужных или повреждённых клеток, поддерживая тканевой гомеостаз. Ежедневно в организме человека образуется и уничтожается десятки миллиардов клеток [1]. Морфологические изменения включают сжатие объёма, конденсацию хроматина, деструкцию ядра и фрагментацию ДНК.
Биохимически апоптоз реализуется через каспазный каскад. Инициирующие каспазы (8, 9, 10) активируют эффекторные каспазы (3, 6, 7), которые разрушают структурные и регуляторные белки клетки. Основные пути активации — рецепторный (внешний) и митохондриальный (внутренний) [3, 1].
Регуляция апоптоза осуществляется балансом про- и антиапоптотических белков семейства Bcl-2. Нарушение этого равновесия ведёт к патологиям, включая опухолевую трансформацию и лекарственную устойчивость. Апоптоз также участвует в контроле постмитотической гибели полиплоидных клеток, предотвращая геномную нестабильность [11].
- Молекулярные липидные медиаторы в апоптозе
Церамид — центральный липидный медиатор, запускающий проапоптотические сигналы через активацию протеинкиназ (PKC, тирозинкиназ) [1, 5]. Он модифицирует активность ионных каналов, особенно калиевых (K⁺). N-ацил-сфингозин, производное церамида, вызывает инактивацию K⁺-каналов, предположительно через фосфорилирование тирозинкиназами [10]. Блокада K⁺-каналов ведёт к деполяризации мембраны, что создаёт условия для апоптоза. Аналогичные процессы происходят на митохондриальной мембране, способствуя потере потенциала [10].
Церамид повышает внутриклеточный кальций и активные формы кислорода, усиливая проапоптотические реакции [5, 4]. Как вторичный мессенджер, он связывает активацию киназ с изменением электрофизиологических характеристик клетки. Избыточное накопление церамидов наблюдается при инсулинорезистентности и диабете 2 типа, что указывает на его роль в метаболических нарушениях [4, 2].
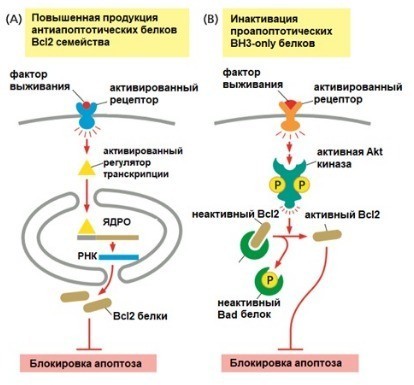
Рисунок 1 — Схемы молекулярных липидных медиаторов и путей передачи сигнала в апоптозе
- Каспазный каскад: ключевой механизм реализации апоптоза
Каспазы — семейство цистеиновых протеаз, расщепляющих субстраты после аспартата. Они играют ключевую роль в апоптозе, а также участвуют в воспалении и некрозе [8]. Все каспазы синтезируются в неактивной форме (зимогены) и активируются в составе белковых комплексов (апоптосомы, DISC, PIDD-осомы) [1, 3].
Выделяют три группы:
- Инициаторные каспазы (8, 9) — активируют эффекторные каспазы.
- Эффекторные каспазы (3, 7) — расщепляют структурные и регуляторные белки (iCAD, ROCK-1, гельзолин), вызывая фрагментацию ДНК и морфологические изменения [1, 5].
- Воспалительные каспазы — активируют провоспалительные цитокины.
Активация каспаз тесно связана с сигналами апоптоза: выход цитохрома c из митохондрий запускает апоптосому и каспазу-9, а внешние сигналы через DISC активируют каспазу-8. Каспазы расщепляют сотни белков, обеспечивая необратимую гибель клетки. Их ингибирование блокирует апоптоз, что имеет терапевтический потенциал [3].
5.Внешний путь активации апоптоза
Внешний путь инициируется связыванием внеклеточных лигандов (FasL, TNF, Apo2L) с рецепторами смерти (CD95, TNFR1, DR3–5), имеющими цитоплазматический домен смерти (DD) [3, 4]. Тримеризация рецепторов приводит к рекрутированию адаптеров (FADD для CD95, TRADD для TNFR1) и формированию DISC (death-inducing signaling complex). DISC активирует инициаторные каспазы-8/10, которые затем запускают каспазный каскад [2, 3].
Внешний путь обеспечивает быстрый ответ, важный для иммунного надзора и удаления повреждённых клеток. В отличие от него, внутренний путь активируется внутриклеточными стрессорами и вовлекает митохондрии.
- Внутренний (митохондриальный) путь активации апоптоза
Внутренний путь контролируется белками семейства Bcl-2, регулирующими проницаемость внешней мембраны митохондрий. Антиапоптотические белки (Bcl-2, Bcl-xL) препятствуют гибели клетки, проапоптотические (Bax, Bak) — способствуют [2]. Под действием проапоптотических сигналов активируются BH3-only белки (Bim, Puma), которые стимулируют олигомеризацию Bax/Bak и формирование пор (пермеабилизация внешней мембраны митохондрий, ПВММ) [5]. Через поры выходит цитохром c, который в цитоплазме связывается с APAF-1, формирует апоптосому и активирует каспазу-9, запуская каспазный каскад [2, 4].
Антиапоптотические белки связывают и нейтрализуют проапоптотические, предотвращая выход цитохрома c. Многие опухолевые клетки используют гиперэкспрессию Bcl-2 для выживания [7]. Селективные ингибиторы Bcl-2 (BH3-миметики, например венетоклакс) восстанавливают апоптоз в злокачественных клетках, однако резистентность остаётся проблемой [1, 3, 7].
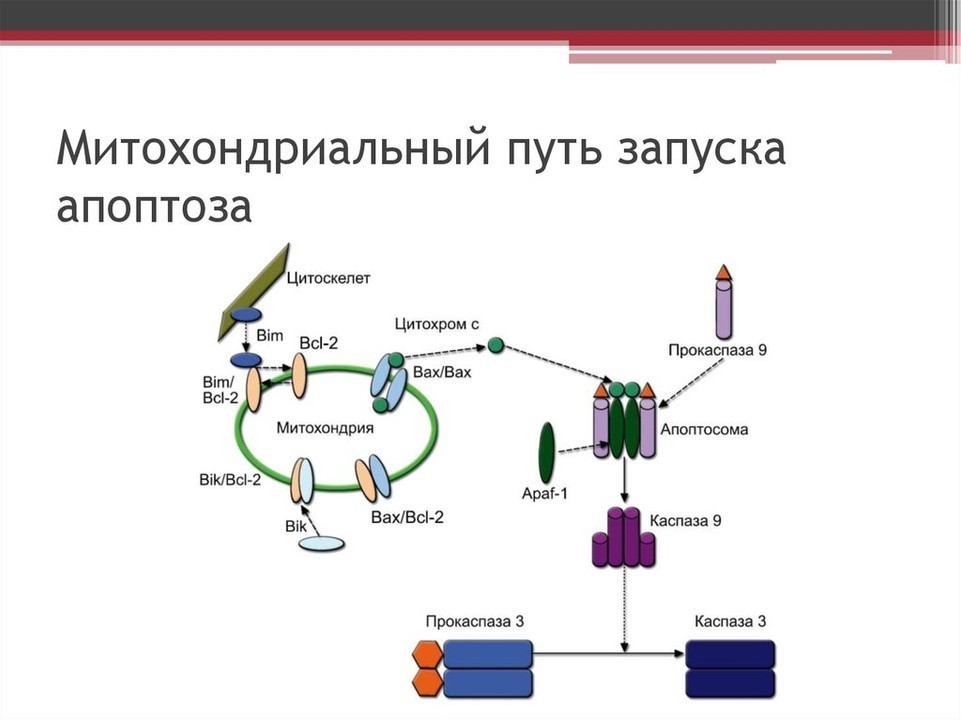
Рисунок 2 — Схемы молекулярных компонентов и взаимодействий внутреннего (митохондриального) пути активации апоптоза
Следующим этапом является рассмотрение механизмов взаимодействия и координации внутреннего и внешнего путей для обеспечения комплексного и адаптивного ответа клетки на проапоптотические сигналы.
7.Модуляция ионных каналов в процессе апоптоза
Ионные каналы, особенно калиевые, модулируют мембранный потенциал и участвуют в регуляции апоптоза. Липидные медиаторы (N-ацил-сфингозин, сфингозин-1-фосфат) изменяют их функцию, обеспечивая дополнительный уровень контроля. Например, блокада калиевых каналов Kv1.3 в лимфоцитах вызывает деполяризацию и апоптоз, что перспективно при хроническом лимфолейкозе [6]. Сфингозин-1-фосфат через рецепторы S1P1–5 регулирует выживание и миграцию клеток; его модуляторы (финголимод) влияют на активность ионных каналов [12].
Изменение активности K⁺-каналов ведёт к деполяризации или гиперполяризации, что служит биофизическим переключателем для апоптоза. Инактивация K⁺-каналов повышает внутриклеточную концентрацию катионов, активируя каспазы [1, 2]. Модуляция ионных каналов через липидные медиаторы дополняет классические сигнальные пути и открывает возможности для таргетной терапии аутоиммунных и онкологических заболеваний [1, 3].
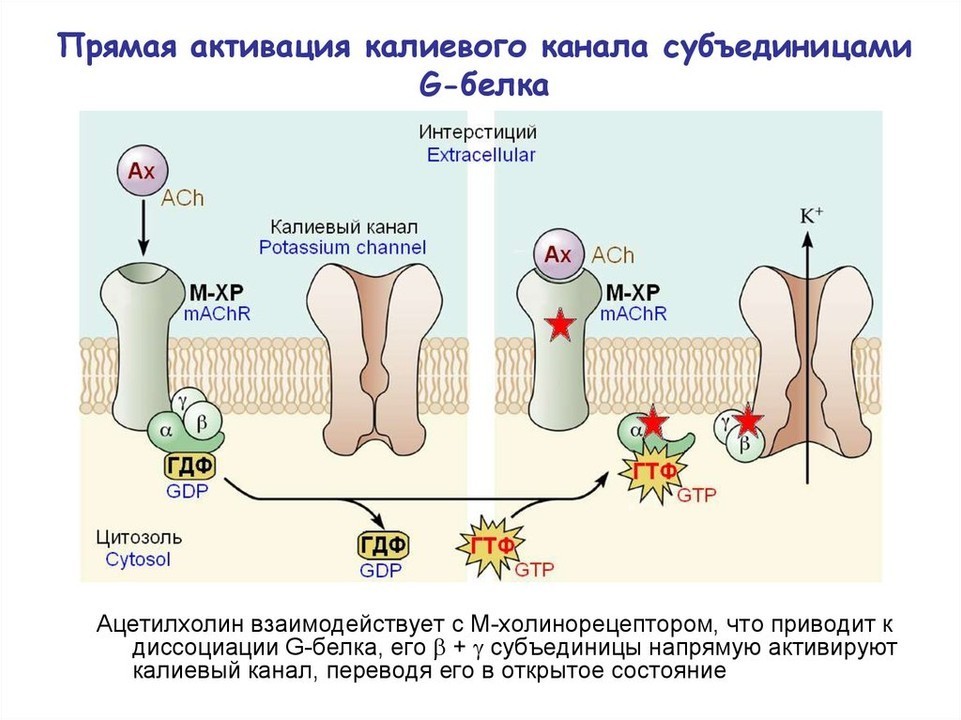
Рисунок 3 — Схемы и описание модуляции ионных каналов в процессе апоптоза
- Сигнальная интеграция: взаимодействие путей активации апоптоза
Ключевым посредником между внешним и внутренним путями является белок Bid. Каспаза-8, активированная в DISC, расщепляет Bid, и его фрагмент транслоцируется к митохондриям, вызывая высвобождение цитохрома c и усиление митохондриального пути [3, 5]. Это позволяет внешним сигналам подключать внутренний каскад.
Сигналы от рецепторов TNF-семейства могут также активировать транскрипционный фактор NF-κB, который индуцирует экспрессию ингибиторов апоптоза (IAP), создавая отрицательную обратную связь и балансируя решение клетки между смертью и выживанием [9].
Внутренний путь дополнительно регулируется фактором p53, который при повреждении ДНК стимулирует экспрессию проапоптотических белков Bcl-2, усиливая митохондриальный выход факторов смерти [1, 5]. Альтернативные механизмы, такие как гранзим В, активируют каспазы независимо от классических путей при иммунном надзоре [10].
Таким образом, интеграция сигналов на уровне прокаспаз, Bcl-2 и транскрипционных факторов обеспечивает адаптивную гибель клеток. Понимание этой сети необходимо для разработки эффективной терапии онкологических, аутоиммунных и нейродегенеративных заболеваний [1–3, 5].
Заключение
Проведённый анализ молекулярных механизмов апоптоза выявил сложное перекрёстное взаимодействие сигнальных путей. Ключевая роль липидных медиаторов, особенно церамида, заключается в активации протеинкиназ и модуляции ионных каналов, что создаёт электрофизиологические условия для запуска каспазного каскада. Каспазы, активируемые внешним (рецепторным) и внутренним (митохондриальным) путями, являются центральным исполнительным механизмом клеточной смерти. Интеграция сигналов через Bid, NF-κB и p53 обеспечивает гибкую адаптацию клетки к стрессу.
Модуляция ионных каналов липидными медиаторами дополняет традиционные сигнальные каскады, демонстрируя взаимозависимость биохимических и биофизических процессов. Перспективы дальнейших исследований связаны с изучением динамики апоптоза в различных клеточных контекстах и разработкой методов избирательного воздействия на ключевые узлы сигнализации. Это имеет прямое прикладное значение для создания терапевтических стратегий при онкологических, аутоиммунных и нейродегенеративных заболеваниях.
Представленные материалы формируют научную базу для контроля жизненного цикла клетки и поддержания тканевого гомеостаза, подчёркивая необходимость системного подхода к изучению апоптоза как интегрированной системы, отвечающей за баланс между жизнью и смертью клетки.
References
1. Рыжов С.В., Новиков В.В. Молекулярные механизмы апоптотических процессов // Российский биотерапевтический журнал. 2002. Т. 1. № 3. С. 22–27.2. Животовский Б.Д., Оррениус С. Апоптоз: биохимия и клеточная биология // Молекулярная биология. 2001. Т. 35. № 2. С. 179–189.
3. Senichkin V.V., Prokhorova E.A., Zhivotovsky B.D., Kopeina G.S. Targeting BCL-2 family proteins in cancer therapy // Biochemistry (Moscow). 2020. Vol. 85. No. 10. P. 1465–1480.
4. Шишкин А.М., Морозов С.Г., Корнева Е.А. Роль церамида в регуляции апоптоза // Биологические мембраны. 2018. Т. 35. № 4. С. 243–251.
5. Пальцев М.А., Иванов А.А. Клеточная биология: учебник. М.: Медицина, 2019. 520 с.
6. Прохорова Е.А., Животовский Б.Д., Копейна Г.С. Каспазный каскад: механизмы активации и роль в апоптозе // Успехи молекулярной онкологии. 2021. Т. 8. № 2. С. 8–19.